
Ion channels are pore forming proteins that provide pathways for the controlled movement of ions into and out of cells. Diseases caused by mutations in genes that encode ion channel subunits or regulatory proteins are reffered to as channelopathies.
Cardiac Channelopathies and Sudden Cardiac Death (SCD)
- It is a challenge for the clinicians to manage sudden death as sometimes patient’s only symptoms at the time of this catastrophic event is sudden death itself.
- Cardiac channelopathies are inherited like other channelopathies involving other organ systems.
- Cardiac channelopathies are associated with other groups of disorders, two of them are:
- Cardiomyopathies- one such inherited disorder is arrhythmogenic cardiomyopathy and other two equally important cardiomyopathies are dilated cardiomyopathy and dilated cardiomyopathy.
- Inherited Channelopathies- Long QT syndrome, Short QT syndrome and Brugada syndrome.
- This article will focus of cardiac channelopathies and one cardiomypathy is also discusses i.e., Arrhytmogenic cardiomyopathy.
- All these disorders are chahracterised by arrhythmias causing sudden cardiac death in a structurally normal heart, variable expression and incomplete penetrance.
- Pathogenesis of cardiac channelopthies- The pathology points towards some underlying genes which get mutated and inherited.
- These genes code for some channels in the plasma membrane of the the vardiac myocytes.
- There is discrepancy in the transfer of ions across the plasma membrane which results in interference of cardiac impulse generation and conduction through the cardiac myocytes causing the myocardium prone to lethal arrhythmias.
- Because the patient and the family mambers are asymptomatic but are at a very high risk of sudden cardiac death, it is very important to educate the family members and their screening for the underlying genetic pathologies.
Arrhythmogenesis in cardiac channelopathies:
- The heterogeneity in expression of ion channels results in spatial and temporal heterogeneity in AP duration and configuration between cardiomyocytes located in different cardiac regions thereby generating spatial voltage gradients that are lrge enough to initiate excitation waves from regions with more positive potentials to less positive regions causing triggered activity and re-entry phenomenon which are responsible for arrhythmogenesis.
Cardiac Channelopathies of clinical significance:
- Brugada syndrome (BrS)
- LQTS ( Long QT Syndrome )
- SQTS ( Short QT Syndrome )
- CPVT ( Catecholaminergic polymorphic VT )
- Early repolarisation syndrome
- Short coupled variant of polymorphic VT
- Early repolarisation syndrome
- Idiopathic ventricular fibrillation
Cardiac Channelopathies: Brugada Syndrome ( BrS )
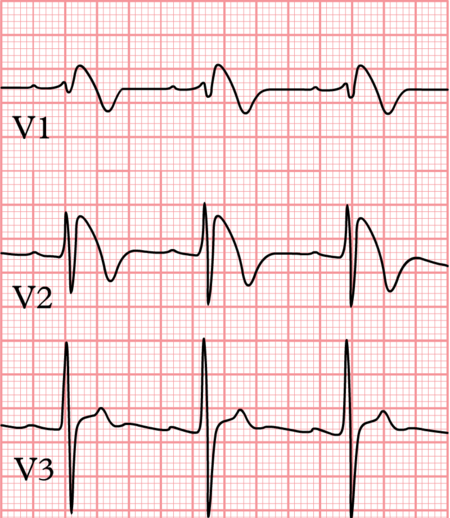
- Brugada Syndrome was described in 1992 as a new clinical entitycharacterized by a typical ECG pattern.
- The imbalance between inwards and outwards ionic currents at phase 1 defines the pathologic substrate for Brugada Syndrome.
- This channelopathy is rare in community and can have varied presentation from asymptomatic state to malignant ventricular tachycardia to sudden cardiac death.
- Typically, this arrhythmic syndrome manifests itself in third to fourth decade with very high male preponderance.
ECG manifestations in Brugada Syndrome:-
ST segment and T wave are typically involved in Brugada Syndrome and pattern of ST-T changes can be any one of the following:-
- Type 1:- Coved ST segment with J point elevation with ST segment elevation >/= 0.2mV followed by negative T wave.
- Type 2:- Saddle back configuration with high takeoff of ST segment >0.2mV followed by downsloping ST elevation ending in positive or biphasic T wave without touching baseline.
- Type 3:- ST segment elevation of <0.1mV with either of the morphologies.
Cardiac Channeloapthies: Long QT Syndrome ( LQTS )
LQTS is a genetic channelopathy that is associated with prolonged ventricular repolarisation manifest electrocardiographically as prolonged QT interval and clinically as increased propensity to syncope, polymorphic ventricular tachycardia ( Torsades de pointes ) and sudden arrhythmic death.
Arrhythmogenesis in LQTS:-
- A number of mutations in the genes encoding for the transmembrane sodium or potassium ion channel proteins can result in delayed repolarisation which is reflected in ECG as prolonged QT interval.
- The initiation of TDP in patients with LQTS is usually dependent on a pause in the electrical activity created by bradycardia or an extrasystole, both of which lead to a longer cycle length.
Diagnosis of LQTS:-
- The diagnosis of LQTS primarily depends on the clinical features, family history, and ECG findings of the patient.
- Scwhartz criteria provide a quantitative approach to the diagnosis of LQTS by allocating numerical points to these three factors and divide the possibility of LQTS into low, intermediate and high probability ranges.
Cardiac Channeloapthies: Short QT Syndrome
- This is a rare channelopathy with high incidence of ventricular fibrillation and sudden cardiac death in affected patients.SQTS has been described as a autosomal dominant channelopathy which results in abbreviation of ventri
- cular repolarisation and increasing the transmural heterogeneity of repolarisation, thus predisposing to ventricular arrhythmia.
- Due to abbreviation of electrical activation, effective refractory period in both atrial and ventricular muscle tissue is low, predisposing to ventricular and atrial fibrillation.
Cardiac Channeloapthies: Catecholaminergic Polymorphic Ventricular Tachycardia
- CPVT is a genetic disease characterized by adrenergically mediated ventricular arrhythmias causing syncope, cardiac arrest and sudden cardiac death in young individuals with structurally normal hearts. Increase in intracytoplasmic calcium ions due to gain of function mutation in RyR2 gene OR loss of function mutation in calsequestrin gene (CaSQ2) leads to delayed after depolarisation and triggered activity.
- Polymorphic ventricular tachycardia may be mediated by re-entry phenomenon. This syndrome usually carries high mortality rates compared to LQTS and is poorly responsive to beta blocker therapy.
- In the article above I have described the Cardiac channelopathies in detail. I hope you visitors have understood the topic well. Any discrepancies are welcome for rectification. Kindly comment below.